
 购物车
购物车


- 首页
- >
- GENESEED
- >
升级服务!单细胞全转录组测序,解锁circRNA的单细胞分辨率视野!
传统Bulk-seq常导致circRNA分析的严重误判!circRNA常表现出高度的细胞或组织特异性,在不同细胞中各司其职。但传统的批量测序(Bulk-seq)只能给出“大杂烩”式的平均结果,不仅掩盖了关键细胞亚群的特有circRNA信号,还可能导致严重的结果误判:① 混合数据“失真”,扭曲circRNA调控关系;②遗漏在特定细胞中差异表达的关键circRNA。
要想看清真相,就必须走进每一个“单细胞”。但遗憾的是,市面上主流的单细胞测序平台,大多通过捕获带有poly(A)尾的RNA进行检测,因而主要分析典型的mRNA,几乎检测不到缺乏poly(A)尾的RNA,尤其是circRNA。
单细胞全转录组测序突破性实现特异性表达的circRNA分析!吉赛生物重磅推出单细胞全转录组测序服务,帮助广大科研工作者解决circRNA细胞异质性分析难题,解锁每一个细胞中完整的RNA世界!
吉赛生物单细胞全转录组测序
技术原理
吉赛生物单细胞全转录组测序利用SeekOne®数字液滴仪,基于微流控技术原理,通过油包水实现单细胞分离捕获,利用核酸修饰的凝胶珠(Barcoded Beads)对不同细胞来源的RNA分子进行分子标记,利用随机引物抓取全长范围的mRNA、circRNA及lncRNA等RNA, 并采用blocker技术去除细胞内的 rRNA ,构建高通量测序文库。最后采用质检合格的文库进行测序,然后结合比对数据和代表细胞身份的Barcode信息,进行细胞聚类、差异基因筛选和差异基因功能分析等。
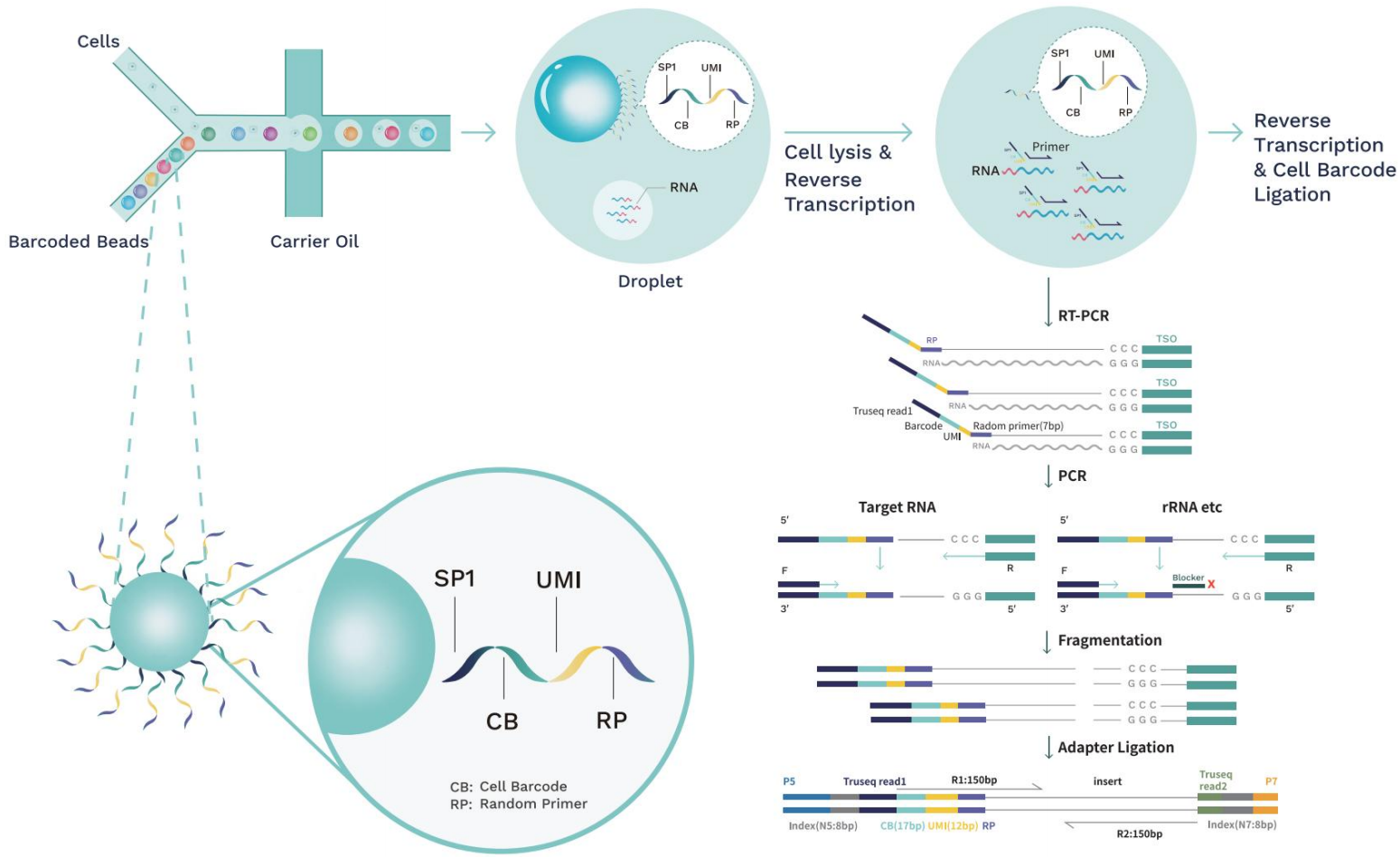
图1 单细胞全转录组测序技术原理。
服务优势
① 突破RNA类型限制,实现无偏差检测
不仅可高效检测mRNA,更覆盖各类非poly(A)转录本:
突破性实现单细胞分辨率下circRNA的精准检测,有利于捕捉在特定细胞中起核心作用的circRNA,探索circRNA的调控网络,挖掘新的circRNA生物标志物和治疗靶点。
同时大幅提升lncRNA检出率,以及实现微生物(如病毒、原核生物)来源的non-poly(A)基因检测,真实还原样本转录组的全貌,有利于更精准区分细胞亚型、捕捉实验样本的细微动态变化。
② 覆盖全序列,释放分析潜能
随机引物捕获,打破3’或5’端转录组的局限,同时利用Read1与Read2两端序列信息,实现全序列覆盖,确保每一条测序读段均有效用于分析,为可变剪接分析、突变检测、新转录本发现等研究提供更全面、可靠的数据支撑。
③ 显著提升灵敏度,无惧降解挑战
即使冻存或短时甲醛固定RNA发生部分降解,仍可稳定捕获并准确检出,降低漏检率,适用于珍贵或部分降解样本,拓宽研究样本范围。
④ 单细胞分辨率,准确描绘基因表达谱
实现单细胞水平基因突变或全转录组差异表达检测,更准确鉴定驱动差异的细胞来源,探索差异细胞微环境特征,在单细胞层面分析细胞功能改变。
⑤ 特色生信分析,全面分析转录组
除了常规的单细胞测序分析(如细胞聚类分析、细胞类型鉴定、Marker基因分析、功能注释与分析、组间差异分析等),还额外增加针对性的circRNA、lncRNA表达和功能分析,以及变异、可变剪接等分析,提供多维度数据。
性能对比
吉赛单细胞全转录组测序在不同样本中,均能实现单细胞分辨率下circRNA的精准检出,还大幅提高了lncRNA的检出率!
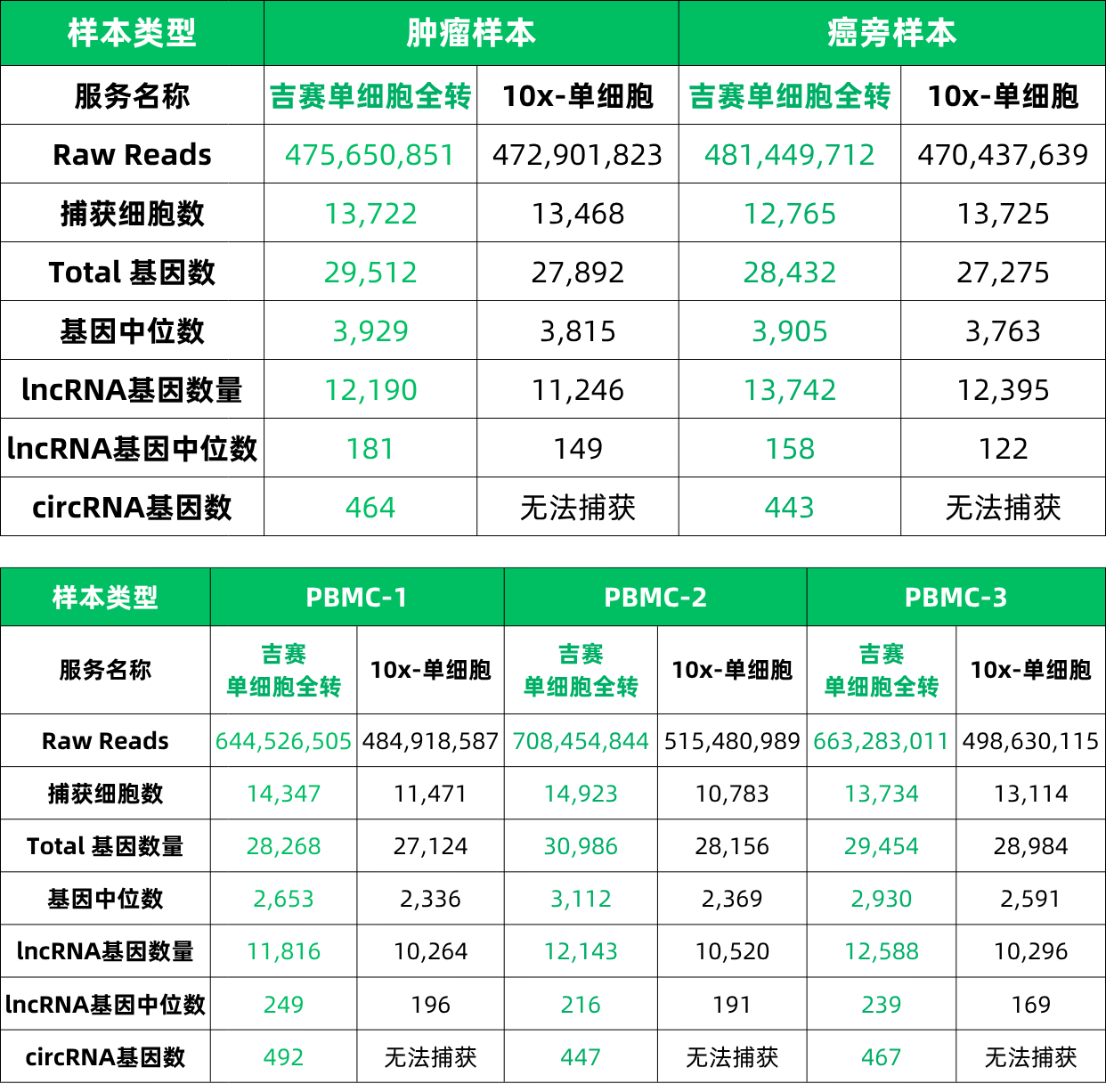
应用案例
(1)研究非编码RNA
论文标题:Assessment of the cardiac noncoding transcriptome by single-cell RNA sequencing identifies FIXER, a conserved profibrogenic long noncoding RNA
发表期刊:Circulation
lncRNA在许多心血管疾病中具有关键功能,它们比编码基因更具有细胞特异性。研究使用单细胞全转录组测序技术对梗死后非心肌细胞中的lncRNA进行了评估,基于lncRNA的表达从心肌成纤维细胞、肌成纤维细胞和心外膜细胞鉴定到7种细胞亚群,再通过细胞亚群表达的mRNA富集的功能模块和文献报道,将这些亚群注释为抑制细胞增殖、细胞活化、血管分化、DNA 修复、有丝分裂、细胞分化等功能相关细胞亚型。研究证明除了传统的mRNA表达模式外,也可以通过lncRNA表达水平识别出哺乳动物心脏中的各种细胞类型,为lncRNA的深入研究开拓了新的思路。[1]
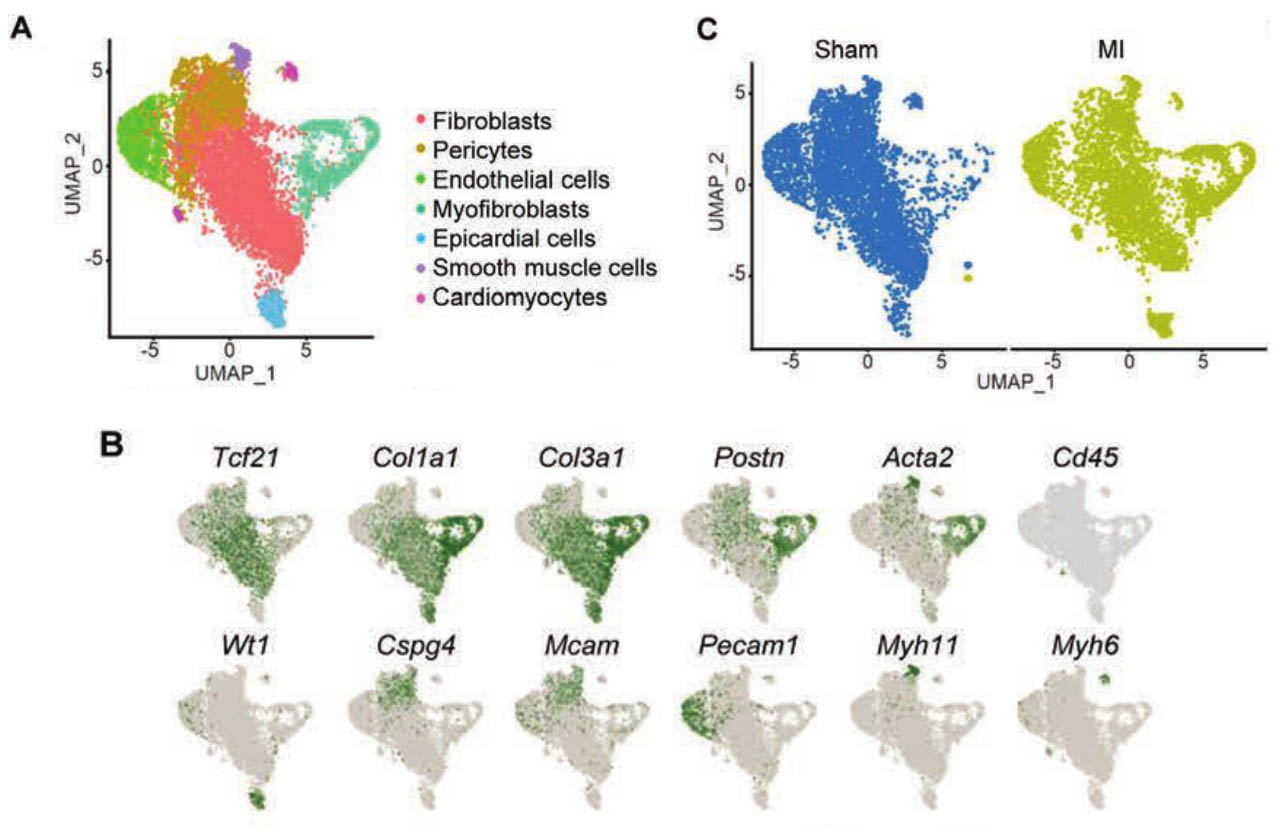
图2 通过单细胞RNA测序确定相关的心肌细胞类型的lncRNA表达谱。A:完全基于lncRNA的表达的非心肌细胞UMAP。B:UMAP图显示每个簇的细胞类型特异性标记的表达。C:UMAP表明假手术和梗死心脏中的细胞簇。
(2)分析可变剪接
论文标题:Splicing diversity enhances the molecular classification of pituitary neuroendocrine tumors
发表期刊:Nat Commun
研究通过联合Bulk-seq和单细胞全转录组测序,全面研究垂体神经内分泌肿瘤(PitNET)表达谱中的可变剪接(AS)失调,揭示了单细胞分辨率下的剪接异质性,以及不同肿瘤细胞类型的剪接失调,还有效地区分了沉默的促肾上腺皮质激素亚型,并定义了一种与更差的临床表现和ESRP1表达变化驱动剪接异常有关的独特TPIT谱系亚型。总之,研究表征了PitNET中亚型特异性AS概况,增强了对PitNET亚型的理解。[2]
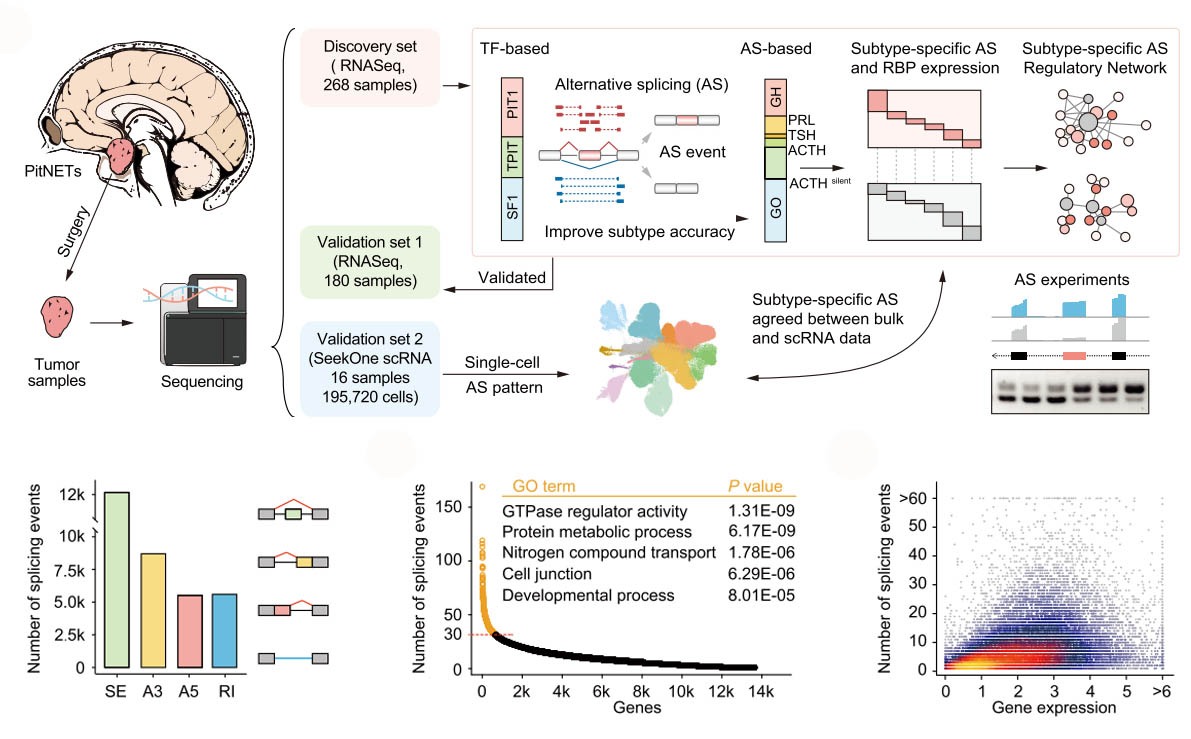
图3 单细胞全转录组测序展现PitNet的剪接多样性。
(3)探索细胞异质性和状态变化
论文标题:Single-cell nascent transcription reveals sparse genome usage and plasticity
发表期刊:Cell
研究利用生物素联合5-EU对新生RNA进行标记,再结合单细胞全转录组技术,基于随机引物无偏好地捕获编码与非编码RNA的优势,以极高的灵敏度和全基因组覆盖范围捕获新生和成熟转录组,呈现了mRNA和基因间非编码RNA 的不同动力学特征,提供了一个独特且全面的单细胞转录动态和异质性的视图。[3]
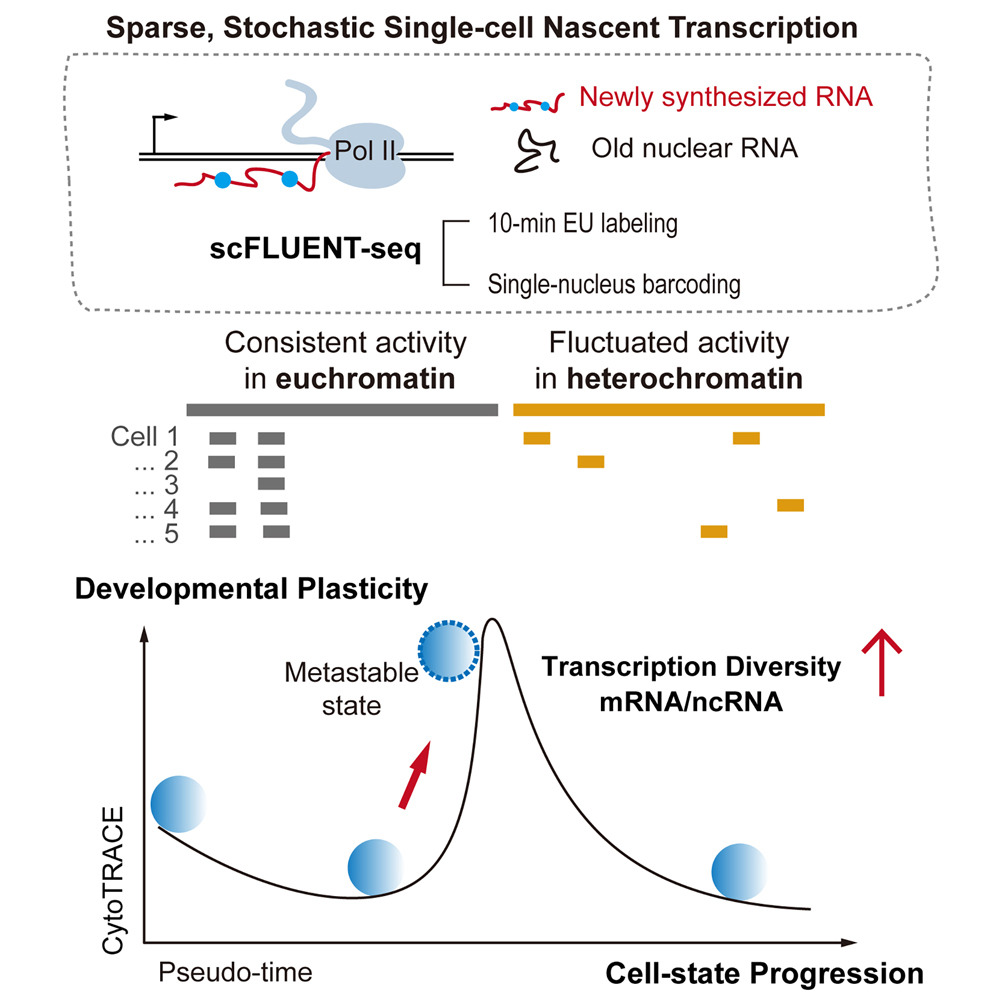
图4 单细胞全转录组测序方案。
(4)癌细胞基因突变研究
论文标题:Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer
发表期刊:Nat Genet
研究使用单细胞/核转录组(Sc/SnRNA-seq)、空间转录组学、细胞成像、Bulk RNA-seq、WES、蛋白组、磷酸化修饰组等多组学技术,分析31例胰腺导管腺癌(PDAC)患者的83个样本,揭示了胰腺癌发生过程中,从正常细胞到癌变前细胞,从癌变前细胞向胰腺癌细胞转变的细节信息。研究将突变映射到单个细胞上,通过比较肿瘤细胞的每个亚群与给定的KRAS突变的基因表达谱来检测KRAS突变的影响,结果表明在同一患者中携带不同KRAS驱动突变的两个不同的克隆具有不同的基因表达谱。[4]
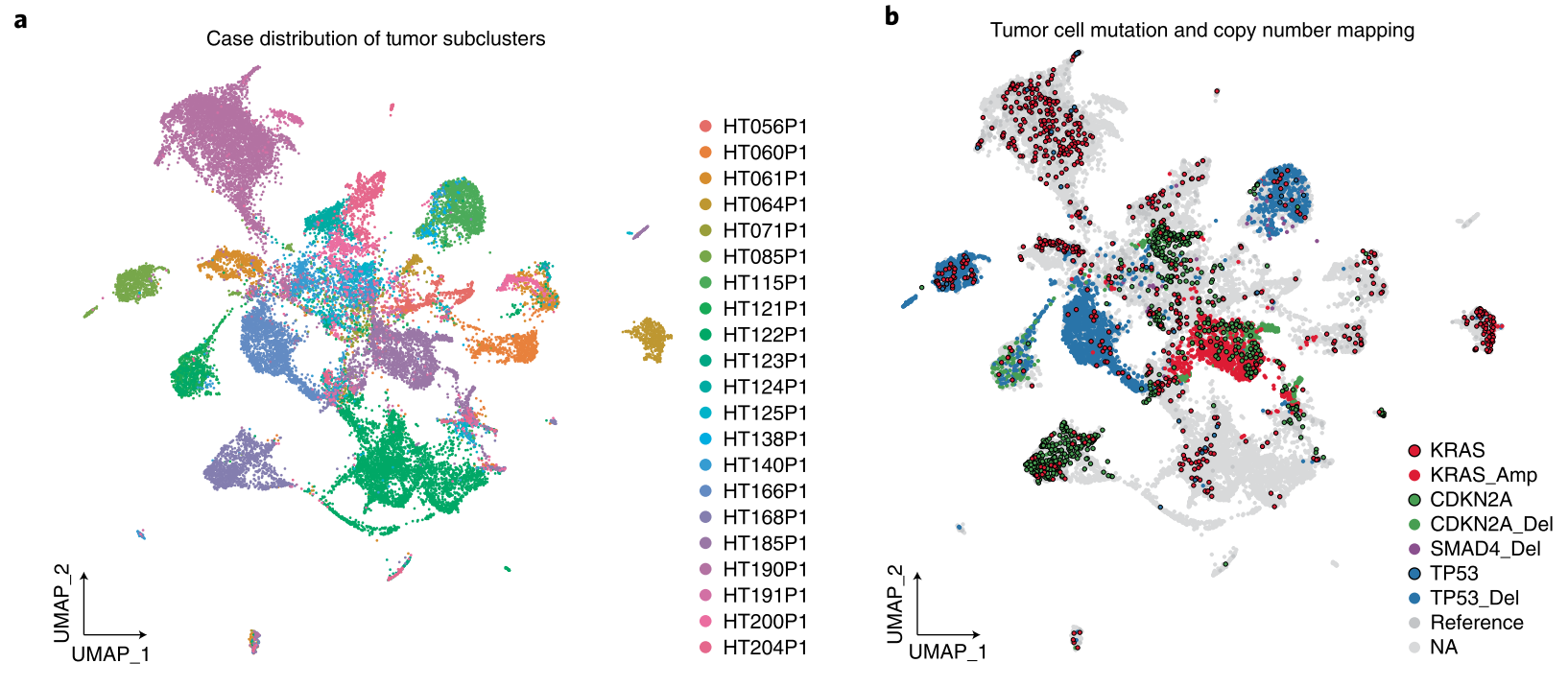
图5 PDAC中的KRAS突变信号。a:用病例ID标记的肿瘤细胞簇;b:用基因组改变(突变和拷贝数变化)标记肿瘤细胞。
参考资料
[1]Aghagolzadeh P, et al. Assessment of the Cardiac Noncoding Transcriptome by Single-Cell RNA Sequencing Identifies FIXER, a Conserved Profibrogenic Long Noncoding RNA. Circulation. 2023;148(9):778-797.
[2]Huang Y, et al. Splicing diversity enhances the molecular classification of pituitary neuroendocrine tumors. Nat Commun. 2025;16(1):1552.
[3]Ma S, et al. Single-cell nascent transcription reveals sparse genome usage and plasticity. Cell. 2025:S0092-8674(25)01034-7.
[4]Cui Zhou D, et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat Genet. 2022;54(9):1390-1405.

 广州市黄埔区开源大道11号科技企业加速器A区6栋2楼
广州市黄埔区开源大道11号科技企业加速器A区6栋2楼


 geneseed@geneseed.com.cn
geneseed@geneseed.com.cn